Update: see detailed comments and analysis here and here by Sasha Gusev. Gusev claims that the problems identified in Figs 4,7 are the result of incorrect calculation of the SE (4) and failure to exclude related individuals in the Framingham data (7).
Bioinformaticist E. Stovner asked about a recent PNAS paper which is critical of GCTA. My comments are below.
It's a shame that we don't have a better online platform (e.g., like Quora or StackOverflow) for discussing scientific papers. This would allow the authors of a paper to communicate directly with interested readers, immediately after the paper appears. If the authors of this paper want to correct my misunderstandings, they are welcome to comment here!
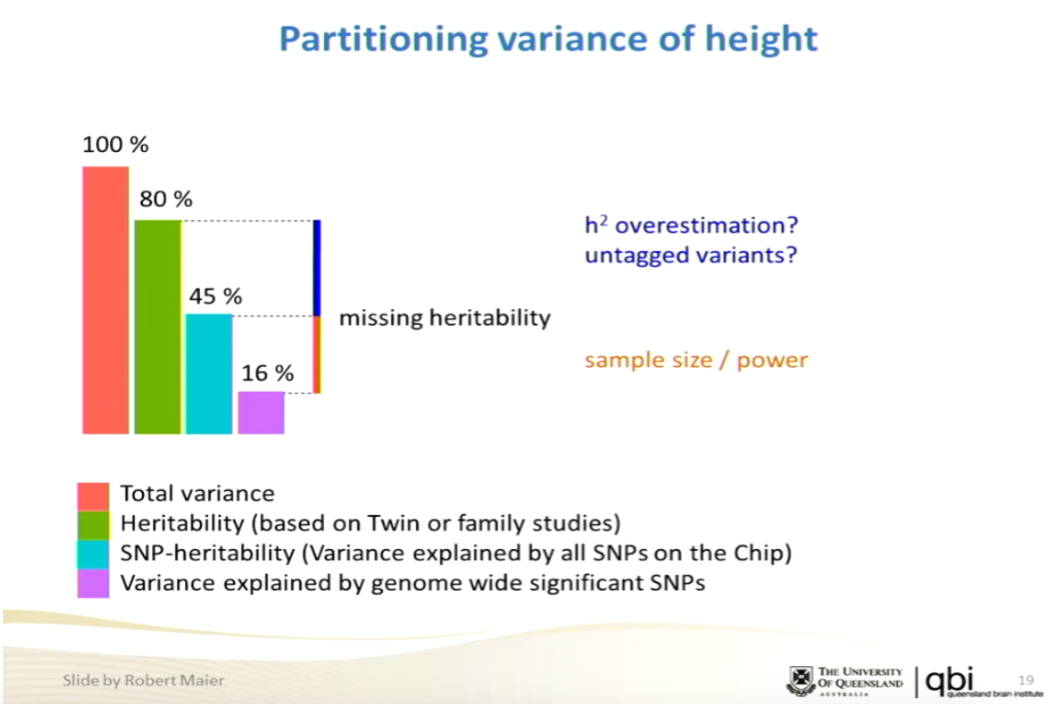
I took a quick look at it. My guess is that Visscher et al. will respond to the paper. It has not changed my opinion of GCTA. Note I have always thought the standard errors quoted for GCTA are too optimistic, as the method makes strong assumptions (e.g., fits a model with Gaussian random effect sizes). But directionally it is obvious that total h2 accounted for by all common SNPs is much larger than what you get from only genome wide significant hits obtained in early studies. For example, the number of genome wide significant hits for some traits (e.g., height) has been growing steadily, along with h2 accounted for using just those hits, eventually approaching the GCTA prediction. That is, even *without* GCTA the steady progress of GWAS shows that common variants account for significant heritability (amount of "missing" heritability steadily declines with GWAS sample size), so the precise reliability of GCTA becomes less important.Here is the paper:
Regarding this paper, they make what sound like strong theoretical points in the text, but the simulation results don't seem to justify the aggressive rhetoric. The only point they really make in figs 4,7 is that the error estimate from GCTA in the case where SNP coverage is inadequate (i.e., using 5k out of 50k SNPs) are way off. But this doesn't correspond to any real world study that we care about. Real world results show that as you approach ~few x 100k SNPs used the h2 result asymptotes (approaches its limiting value), because you have enough coverage of common variants. The authors of the paper seem confused about this point -- see "Saturation of heritability estimates" section.
What they should do is simulate repeatedly with multiple disjoint populations (using good SNP coverage) and see how the heritability results fluctuate. But I think that kind of calculation has been done by other people and does not show large fluctuations in h2.
Well, since you got me to write this much already I suppose I should promote this to an actual blog post at some point ... Please keep in mind that I've only given the paper a quick read so I might be missing something important. Happy New Year!
Limitations of GCTA as a solution to the missing heritability problemIt's important to note that although GCTA fits a model with random effects, it purports to estimate the heritability of more realistic genetic architectures with some other (e.g., sparse) distribution of effect sizes (see Lee and Chow paper at bottom of this post). The authors of this PNAS paper seem to take the random effects assumption more seriously than the GCTA originators themselves. The latter fully expected a saturation effect once enough SNPs are used; the former seem to think it violates the fundamental nature of the model. Indeed, AFAICT, the toy models in the PNAS simulations assume all 50k SNPs affect the trait, and they run simulations where only 5k at a time are included in the computation. This is likely the opposite of the real world situation, in which a relatively small number (e.g., ~10k SNPs) affect the trait, and by using a decent array with > 200k SNPs one already obtains sensitivity to the small subset.
http://www.pnas.org/content/early/2015/12/17/1520109113
The genetic contribution to a phenotype is frequently measured by heritability, the fraction of trait variation explained by genetic differences. Hundreds of publications have found DNA polymorphisms that are statistically associated with diseases or quantitative traits [genome-wide association studies (GWASs)]. Genome-wide complex trait analysis (GCTA), a recent method of analyzing such data, finds high heritabilities for such phenotypes. We analyze GCTA and show that the heritability estimates it produces are highly sensitive to the structure of the genetic relatedness matrix, to the sampling of phenotypes and subjects, and to the accuracy of phenotype measurements. Plausible modifications of the method aimed at increasing stability yield much smaller heritabilities. It is essential to reevaluate the many published heritability estimates based on GCTA.
One can easily show that genetic architectures of complex traits tend to be sparse: most of the heritability is accounted for by a small subset of alleles. (Here "small" means a small fraction of ~ millions of SNPs: e.g., 10k SNPs.) See section 3.2 of On the genetic architecture of intelligence and other quantitative traits for an explanation of how to roughly estimate the sparsity using genetic Hamming distances. In our work on Compressed Sensing applied to genomics, we showed that much of the heritability for many complex traits can be recovered if sample sizes of order millions are available for analysis. Once these large data sets are available, this entire debate about missing heritability and GCTA heritability estimates will recede in importance. (See talk and slides here.)
For more discussion, see Why does GCTA work?
This paper, by two of my collaborators, examines the validity of a recently introduced technique called GCTA (Genome-wide Complex Trait Analysis). GCTA allows an estimation of heritability due to common SNPs using relatively small sample sizes (e.g., a few thousand genotype-phenotype pairs). The new method is independent of, but delivers results consistent with, "classical" methods such as twin and adoption studies. To oversimplify, it examines pairs of unrelated individuals and computes the correlation between pairwise phenotype similarity and genotype similarity (relatedness). It has been applied to height, intelligence, and many medical and psychiatric conditions.
When the original GCTA paper (Common SNPs explain a large proportion of the heritability for human height) appeared in Nature Genetics it stimulated quite a lot of attention. But I was always uncertain of the theoretical justification for the technique -- what are the necessary conditions for it to work? What are conservative error estimates for the derived heritability? My impression, from talking to some of the authors, is that they had a mainly empirical view of these questions. The paper below elaborates significantly on the theory behind GCTA.
Conditions for the validity of SNP-based heritability estimation
James J Lee, Carson C Chow
doi: 10.1101/003160
...