
A recent PNAS paper by researchers at Stanford claims to identify many problems with GCTA. The conclusions of this paper have been hotly contested by the GCTA authors and others.
Earlier post (January 1, 2016) on PNAS paper Limitations of GCTA as a solution to the missing heritability problem. (See also: many posts on this blog which mention GCTA.)This dispute shows the utility of blogs (Gusev) and biorxiv for rapid scientific discussion. Some of the commentaries listed above are 20+ pages long with figures and equations. This discussion would not have been possible (or would have taken months or years) in a journal setting.
Detailed comments and analysis here and here by Sasha Gusev. Gusev claims that the problems identified in Figs 4,7 are the result of incorrect calculation of the SE (4) and failure to exclude related individuals in the Framingham data (7).
GCTA authors Visscher, Yang, et al. respond to PNAS paper -- they accept none of the criticisms (February 13, 2016 biorxiv).
PNAS authors reply to Visscher, Yang, et al. comments (February 16, 2016 bioarxiv). They claim that relatedness thresholding used with GCTA analysis is flawed and that residual standard errors are much larger than claimed.
Gamazon and Park (February 18, 2016 bioarxiv) question spectral analysis and random matrix theory results in the PNAS paper. (I believe this is the first critique which looks at the mathematics of the PNAS paper, as opposed to simulation results.)
The next step should be a mini-workshop conducted online, with each group allowed 30 min to present their results, followed by questions :-)
I've always felt that the real weakness of GCTA is the assumption of random effects. A consequence of this assumption is that if the true causal variants are atypical (e.g., in terms of linkage disequilibrium) among common SNPs, the results could be biased. It is impossible to evaluate this uncertainty at the moment because we do not yet know the genetic architectures of any complex traits. See Why does GCTA work? for more discussion and a link to work by Lee and Chow examining this issue.
Recently, a promising new method (Heritability Estimates from Summary Statistics) has been proposed which does not make assumptions about the effect size distribution -- it uses GWAS estimates of effect size to directly estimate variance accounted for by each region of the genome. The initial application of this method also suggests significant heritability due to common variants.
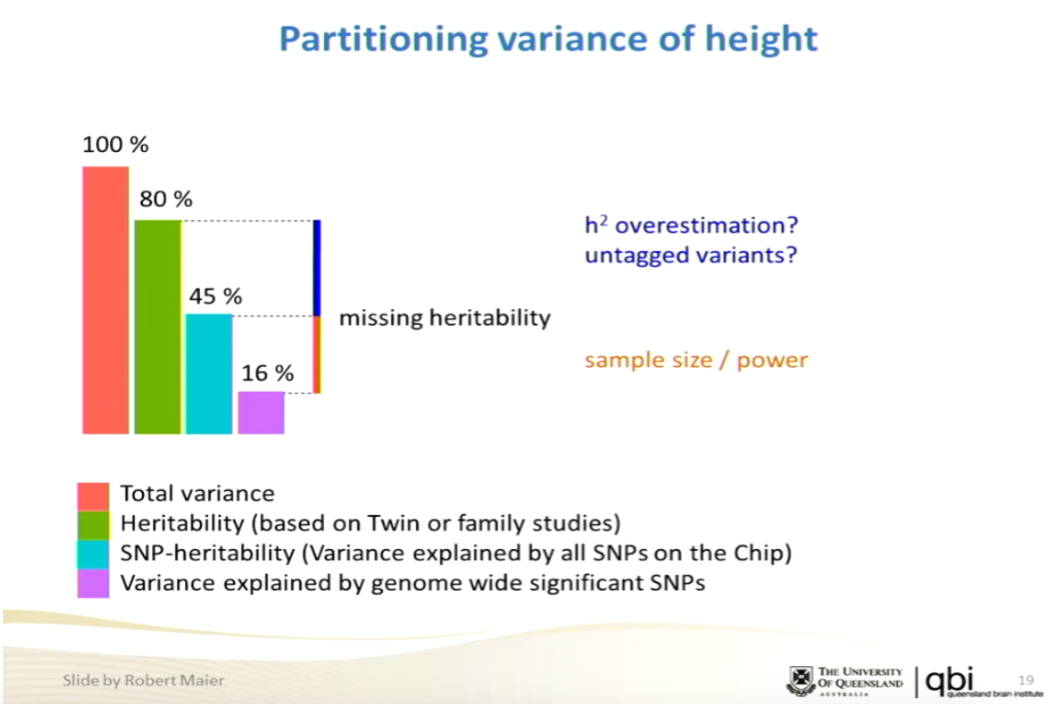
The broader debate over whether common variants will eventually account for significant heritability in many complex traits has been going on for years now. The centrality of GCTA results to this question decreases by the year as more and more heritability is accounted for by specific loci identified at genome wide significance in well-powered GWAS. For example, this slide (see talk on genomic prediction I gave in 2015 at NIH and HLI) shows that GWAS hits on height now account for 16% of total variance. That means a predictor could be constructed with correlation ~0.4 to the actual trait. I think the argument is basically over, unless you have some ulterior motive for denying the potential of genomic prediction.
No comments:
Post a Comment