
Thursday, June 27, 2019
Manifold Podcast #13: Joe Cesario on Political Bias and Problematic Research Methods in Social Psychology
Corey and Steve continue their discussion with Joe Cesario and examine methodological biases in the design and conduct of experiments in social psychology and ideological bias in the interpretation of the findings. Joe argues that experiments in his field are designed to be simple, but that in making experimental set ups simple researchers remove critical factors that actually matter for a police officer making a decision in the real world. In consequence, he argues that the results cannot be taken to show anything about actual police behavior. Joe maintains that social psychology as a whole is biased toward the left politically and that this affects how courses are taught and research conducted. Steve points out the university faculty on the whole tend to be shifted left relative to the general population. Joe, Corey, and Steve discuss the current ideological situation on campus and how it can be alienating for students from conservative backgrounds.
Joseph Cesario's Lab
https://www.cesariolab.com/
Transcript
https://manifoldlearning.com/2019/06/27/episode-013-transcript/
man·i·fold /ˈmanəˌfōld/ many and various.
In mathematics, a manifold is a topological space that locally resembles Euclidean space near each point.
Steve Hsu and Corey Washington have been friends for almost 30 years, and between them hold PhDs in Neuroscience, Philosophy, and Theoretical Physics. Join them for wide ranging and unfiltered conversations with leading writers, scientists, technologists, academics, entrepreneurs, investors, and more.
Steve Hsu is VP for Research and Professor of Theoretical Physics at Michigan State University. He is also a researcher in computational genomics and founder of several Silicon Valley startups, ranging from information security to biotech. Educated at Caltech and Berkeley, he was a Harvard Junior Fellow and held faculty positions at Yale and the University of Oregon before joining MSU.
Corey Washington is Director of Analytics in the Office of Research and Innovation at Michigan State University. He was educated at Amherst College and MIT before receiving a PhD in Philosophy from Stanford and a PhD in a Neuroscience from Columbia. He held faculty positions at the University Washington and the University of Maryland. Prior to MSU, Corey worked as a biotech consultant and is founder of a medical diagnostics startup.
Monday, June 24, 2019
Ulam on von Neumann, Godel, and Einstein

Ulam expresses so much in a few sentences! From his memoir, Adventures of a Mathematician. Above: Einstein and Godel. Bottom: von Neumann, Feynman, Ulam.
When it came to other scientists, the person for whom he [vN] had a deep admiration was Kurt Gödel. This was mingled with a feeling of disappointment at not having himself thought of "undecidability." For years Gödel was not a professor at Princeton, merely a visiting fellow, I think it was called. Apparently there was someone on the faculty who was against him and managed to prevent his promotion to a professorship. Johnny would say to me, "How can any of us be called professor when Gödel is not?" ...
As for Gödel, he valued Johnny very highly and was much interested in his views. I believe knowing the importance of his own discovery did not prevent Gödel from a gnawing uncertainty that maybe all he had discovered was another paradox à la Burali Forte or Russell. But it is much, much more. It is a revolutionary discovery which changed both the philosophical and the technical aspects of mathematics.
When we talked about Einstein, Johnny would express the usual admiration for his epochal discoveries which had come to him so effortlessly, for the improbable luck of his formulations, and for his four papers on relativity, on the Brownian motion, and on the photo-electric quantum effect. How implausible it is that the velocity of light should be the same emanating from a moving object, whether it is coming toward you or whether it is receding. But his admiration seemed mixed with some reservations, as if he thought, "Well, here he is, so very great," yet knowing his limitations. He was surprised at Einstein's attitude in his debates with Niels Bohr—at his qualms about quantum theory in general. My own feeling has always been that the last word has not been said and that a new "super quantum theory" might reconcile the different premises.

Saturday, June 22, 2019
Silicon Oligarchs: Winner Take All?

Joel Kotkin is a Presidential Fellow in Urban Futures at Chapman University and Executive Director for the Center for Opportunity Urbanism.
What Do the Oligarchs Have in Mind for Us?
...This tiny sliver of humanity, with their relatively small cadre of financiers, engineers, data scientists, and marketers, now control the exploitation of our personal data, what Alibaba founder, Jack Ma calls the “electricity of the 21st century.” Their “super platforms,” as one analyst noted, “now operate as “digital gatekeepers” lording over “e-monopsonies” that control enormous parts of the economy. Their growing power, notes a recent World Bank Study, is built on “natural monopolies” that adhere to web-based business, and have served to further widen class divides not only in the United States but around the world.
The rulers of the Valley and its Puget Sound doppelganger now account for eight of the 20 wealthiest people on the planet. Seventy percent of the 56 billionaires under 40 live in the state of California, with 12 in San Francisco alone. In 2017, the tech industry, mostly in California, produced 11 new billionaires. The Bay Area has more billionaires on the Forbes 400 list than any metro region other than New York and more millionaires per capita than any other large metropolis.
For an industry once known for competition, the level of concentration is remarkable. Google controls nearly 90 percent of search advertising, Facebook almost 80 percent of mobile social traffic, and Amazon about 75 percent of US e-book sales, and, perhaps most importantly, nearly 40 percent of the world’s “cloud business.” Together, Google and Apple control more than 95 percent of operating software for mobile devices, while Microsoft still accounts for more than 80 percent of the software that runs personal computers around the world.
The wealth generated by these near-monopolies funds the tech oligarchy’s drive to monopolize existing industries such as entertainment, education, and retail, as well as those of the future, such as autonomous cars, drones, space exploration, and most critically, artificial intelligence. Unless checked, they will have accumulated the power to bring about what could best be seen as a “post-human” future, in which society is dominated by artificial intelligence and those who control it.
What Do the Oligarchs Want?
The oligarchs are creating a “a scientific caste system,” not dissimilar to that outlined in Aldous Huxley’s dystopian 1932 novel, Brave New World. Unlike the former masters of the industrial age, they have little use for the labor of middle- and working-class people—they need only their data. Virtually all their human resource emphasis relies on cultivating and retaining a relative handful of tech-savvy operators. “Software,” Bill Gates told Forbes in 2005, “is an IQ business. Microsoft must win the IQ war, or we won’t have a future.”
Perhaps the best insight into the mentality of the tech oligarchy comes from an admirer, researcher Greg Ferenstein, who interviewed 147 digital company founders. The emerging tech world has little place for upward mobility, he found, except for those in the charmed circle at the top of the tech infrastructure; the middle and working classes become, as in feudal times, increasingly marginal.
This reflects their perception of how society will evolve. Ferenstein notes that most oligarchs believe “an increasingly greater share of economic wealth will be generated by a smaller slice of very talented or original people. Everyone else will increasingly subsist on some combination of part-time entrepreneurial ‘gig work’ and government aid.” Such part-time work has been growing rapidly, accounting for roughly 20 percent of the workforce in the US and Europe, and is expected to grow substantially, adds McKinsey. ...
Thursday, June 20, 2019
CRISPR babies: when will the world be ready? (Nature)
This Nature News article gives a nice overview of the current status of CRISPR technology and its potential application in human reproduction. As we discussed in this bioethics conversation (Manifold Podcast #9 with philosopher Sam Kerstein of the University of Maryland), it is somewhat challenging to come up with examples where gene editing is favored over embryo selection (a well-established technology) for avoidance of a disease-linked mutation.


Nature: ... He found out about a process called preimplantation genetic diagnosis or PGD. By conceiving through in vitro fertilization (IVF) and screening the embryos, Carroll and his wife could all but eliminate the chance of passing on the mutation. They decided to give it a shot, and had twins free of the Huntington’s mutation in 2006.
Now Carroll is a researcher at Western Washington University in Bellingham, where he uses another technique that might help couples in his position: CRISPR gene editing. He has been using the powerful tool to tweak expression of the gene responsible for Huntington’s disease in mouse cells. Because it is caused by a single gene and is so devastating, Huntington’s is sometimes held up as an example of a condition in which gene editing a human embryo — controversial because it would cause changes that would be inherited by future generations — could be really powerful. But the prospect of using CRISPR to alter the gene in human embryos still worries Carroll. “That’s a big red line,” he says. “I get that people want to go over it — I do, too. But we have to be super humble about this stuff.” There could be many unintended consequences, both for the health of individuals and for society. It would take decades of research, he says, before the technology could be used safely.


Thursday, June 13, 2019
Manifold Episode #12: James Cham on Venture Capital, Risk Taking, and the Future Impacts of AI
Manifold Show Page YouTube Channel
James Cham is a partner at Bloomberg Beta, a venture capital firm focused on the future of work. James invests in companies applying machine intelligence to businesses and society. Prior to Bloomberg Beta, James was a Principal at Trinity Ventures and a VP at Bessemer Venture Partners. He was educated in computer science at Harvard and at the MIT Sloan School of Business.
James Cham
https://www.linkedin.com/in/jcham/
Bloomberg Beta
https://www.bloombergbeta.com/
man·i·fold /ˈmanəˌfōld/ many and various.
In mathematics, a manifold is a topological space that locally resembles Euclidean space near each point.
Steve Hsu and Corey Washington have been friends for almost 30 years, and between them hold PhDs in Neuroscience, Philosophy, and Theoretical Physics. Join them for wide ranging and unfiltered conversations with leading writers, scientists, technologists, academics, entrepreneurs, investors, and more.
Steve Hsu is VP for Research and Professor of Theoretical Physics at Michigan State University. He is also a researcher in computational genomics and founder of several Silicon Valley startups, ranging from information security to biotech. Educated at Caltech and Berkeley, he was a Harvard Junior Fellow and held faculty positions at Yale and the University of Oregon before joining MSU.
Corey Washington is Director of Analytics in the Office of Research and Innovation at Michigan State University. He was educated at Amherst College and MIT before receiving a PhD in Philosophy from Stanford and a PhD in a Neuroscience from Columbia. He held faculty positions at the University Washington and the University of Maryland. Prior to MSU, Corey worked as a biotech consultant and is founder of a medical diagnostics startup.
Validation of Polygenic Risk Scores for Coronary Artery Disease in French Canadians

This study reports a validation of Polygenic Risk Scores for Coronary Artery Disease in a French Canadian population. Outliers in PRS are much more likely to have CAD than typical individuals.
In our replication tests of a variety of traits (both disease risks and quantitative traits) using European ancestry validation datasets, there is strong consistency in performance of the predictors. (See AUC consistency below.) This suggests that the genomic predictors are robust to differences in environmental conditions and also moderate differences in ethnicity (i.e., within the European population). The results are not brittle, and I believe that widespread clinical applications are coming very soon.
Validation of Genome-wide Polygenic Risk Scores for Coronary Artery Disease in French CanadiansAmerican Heart Association hails potential of PRS:
Florian Wünnemann , Ken Sin Lo , Alexandra Langford-Alevar , David Busseuil , Marie-Pierre Dubé , Jean-Claude Tardif , and Guillaume Lettre
Genomic and Precision Medicine
Abstract
Background: Coronary artery disease (CAD) represents one of the leading causes of morbidity and mortality worldwide. Given the healthcare risks and societal impacts associated with CAD, their clinical management would benefit from improved prevention and prediction tools. Polygenic risk scores (PRS) based on an individual's genome sequence are emerging as potentially powerful biomarkers to predict the risk to develop CAD. Two recently derived genome-wide PRS have shown high specificity and sensitivity to identify CAD cases in European-ancestry participants from the UK Biobank. However, validation of the PRS predictive power and transferability in other populations is now required to support their clinical utility.
Methods: We calculated both PRS (GPSCAD and metaGRSCAD) in French-Canadian individuals from three cohorts totaling 3639 prevalent CAD cases and 7382 controls, and tested their power to predict prevalent, incident and recurrent CAD. We also estimated the impact of the founder French-Canadian familial hypercholesterolemia deletion (LDLR delta > 15kb deletion) on CAD risk in one of these cohorts and used this estimate to calibrate the impact of the PRS.
Results: Our results confirm the ability of both PRS to predict prevalent CAD comparable to the original reports (area under the curve (AUC)=0.72-0.89). Furthermore, the PRS identified about 6-7% of individuals at CAD risk similar to carriers of the LDLR delta > 15kb mutation, consistent with previous estimates. However, the PRS did not perform as well in predicting incident or recurrent CAD (AUC=0.56-0.60), maybe due to confounding because 76% of the participants were on statin treatment. This result suggests that additional work is warranted to better understand how ascertainment biases and study design impact PRS for CAD.
Conclusions: Collectively, our results confirm that novel, genome-wide PRS are able to predict CAD in French-Canadians; with further improvements, this is likely to pave the way towards more targeted strategies to predict and prevent CAD-related adverse events.
"PRSs, built using very large data sets of people with and without heart disease, look for genetic changes in the DNA that influence disease risk, whereas individual genes might have only a small effect on disease predisposition," said Guillaume Lettre, Ph.D., lead author of the study and an associate professor at the Montreal Heart Institute and Université de Montréal in Montreal, Quebec, Canada. "The PRS is like having a snapshot of the whole genetic variation found in one's DNA and can more powerfully predict one's disease risk. Using the score, we can better understand whether someone is at higher or lower risk to develop a heart problem."
Early prediction would benefit prevention, optimal management and treatment strategies for heart disease. Because PRSs are simple and relatively inexpensive, their implementation in the clinical setting holds great promises. For heart disease, early detection could lead to simple yet effective therapeutic interventions such as the use of statins, aspirin or other medications.
... The American Heart Association named the use of polygenic risk scores as one of the biggest advances in heart disease and stroke research in 2018.
Sadly, reaction to these breakthroughs in human genomics will follow the usual pattern:
It's Wrong! Genomes are too complex to decipher, GWAS is a failure, precision medicine is all hype, biology is so ineffably beautiful and incomprehensible, Hey, whaddaya, you're a physicist! ...
It's Trivial! I knew it all along. Of course, everything is heritable to some degree. Well, if you just get enough data...
I did it First! (Please cite my paper...)
Sunday, June 09, 2019
L1 vs Deep Learning in Genomic Prediction
The paper below by some of my MSU colleagues examines the performance of a number of ML algorithms, both linear and nonlinear, including deep neural nets, in genomic prediction across several different species.
When I give talks about prediction of disease risks and complex traits in humans, I am often asked why we are not using fancy (trendy?) methods such as Deep Learning (DL). Instead, we focus on L1 penalization methods ("sparse learning") because 1. the theoretical framework (including theorems providing performance guarantees) is well-developed, and (relatedly) 2. the L1 methods perform as well or better than other methods in our own testing.
The term theoretical framework may seem unusual in ML, which is at the moment largely an empirical subject. Experience in theoretical physics shows that when powerful mathematical results are available, they can be very useful to guide investigation. In the case of sparse learning we can make specific estimates for how much data is required to "solve" a trait -- i.e., capture most of the estimated heritability in the predictor. Five years ago we predicted a threshold of a few hundred thousand genomes for height, and this turned out to be correct. Currently, this kind of performance characterization is not possible for DL or other methods.
What is especially powerful about deep neural nets is that they yield a quasi-convex (or at least reasonably efficient) optimization procedure which can learn high dimensional functions. The class of models is both tractable from a learning/optimization perspective, but also highly expressive. As I wrote here in my ICML notes (see also Elad's work which relates DL to Sparse Learning):
Note, though, that from an information theoretic perspective (see, e.g., any performance theorems in compressed sensing) it is obvious that we will need much more data than we currently have to advance this program. Also, note that Visscher et al.'s recent GCTA work suggests that additive SNP models using rare variants (i.e., extracted from whole genome data), can account for nearly all the expected heritability for height. This implies that the power of nonlinear methods like DL may not yield qualitatively better results than simpler L1 approaches, even in the limit of very large whole genome datasets.


When I give talks about prediction of disease risks and complex traits in humans, I am often asked why we are not using fancy (trendy?) methods such as Deep Learning (DL). Instead, we focus on L1 penalization methods ("sparse learning") because 1. the theoretical framework (including theorems providing performance guarantees) is well-developed, and (relatedly) 2. the L1 methods perform as well or better than other methods in our own testing.
The term theoretical framework may seem unusual in ML, which is at the moment largely an empirical subject. Experience in theoretical physics shows that when powerful mathematical results are available, they can be very useful to guide investigation. In the case of sparse learning we can make specific estimates for how much data is required to "solve" a trait -- i.e., capture most of the estimated heritability in the predictor. Five years ago we predicted a threshold of a few hundred thousand genomes for height, and this turned out to be correct. Currently, this kind of performance characterization is not possible for DL or other methods.
What is especially powerful about deep neural nets is that they yield a quasi-convex (or at least reasonably efficient) optimization procedure which can learn high dimensional functions. The class of models is both tractable from a learning/optimization perspective, but also highly expressive. As I wrote here in my ICML notes (see also Elad's work which relates DL to Sparse Learning):
It may turn out that the problems on which DL works well are precisely those in which the training data (and underlying generative processes) have a hierarchical structure which is sparse, level by level. Layered networks perform a kind of coarse graining (renormalization group flow): first layers filter by feature, subsequent layers by combinations of features, etc. But the whole thing can be understood as products of sparse filters, and the performance under training is described by sparse performance guarantees (ReLU = thresholded penalization?).However, currently in genomic prediction one typically finds that nonlinear interactions are small, which means features more complicated than single SNPs are unnecessary. (In a recent post I discussed a new T1D predictor that makes use of nonlinear haplotype interaction effects, but even there the effects are not large.) Eventually I expect this situation to change -- when we have enough whole genomes to work with, a DL approach which can (automatically) identify important features (motifs?) may allow us to go beyond SNPs and simple linear models.
Note, though, that from an information theoretic perspective (see, e.g., any performance theorems in compressed sensing) it is obvious that we will need much more data than we currently have to advance this program. Also, note that Visscher et al.'s recent GCTA work suggests that additive SNP models using rare variants (i.e., extracted from whole genome data), can account for nearly all the expected heritability for height. This implies that the power of nonlinear methods like DL may not yield qualitatively better results than simpler L1 approaches, even in the limit of very large whole genome datasets.
Benchmarking algorithms for genomic prediction of complex traits
Christina B. Azodi, Andrew McCarren, Mark Roantree, Gustavo de los Campos, Shin-Han Shiu
The usefulness of Genomic Prediction (GP) in crop and livestock breeding programs has led to efforts to develop new and improved GP approaches including non-linear algorithm, such as artificial neural networks (ANN) (i.e. deep learning) and gradient tree boosting. However, the performance of these algorithms has not been compared in a systematic manner using a wide range of GP datasets and models. Using data of 18 traits across six plant species with different marker densities and training population sizes, we compared the performance of six linear and five non-linear algorithms, including ANNs. First, we found that hyperparameter selection was critical for all non-linear algorithms and that feature selection prior to model training was necessary for ANNs when the markers greatly outnumbered the number of training lines. Across all species and trait combinations, no one algorithm performed best, however predictions based on a combination of results from multiple GP algorithms (i.e. ensemble predictions) performed consistently well. While linear and non-linear algorithms performed best for a similar number of traits, the performance of non-linear algorithms vary more between traits than that of linear algorithms. Although ANNs did not perform best for any trait, we identified strategies (i.e. feature selection, seeded starting weights) that boosted their performance near the level of other algorithms. These results, together with the fact that even small improvements in GP performance could accumulate into large genetic gains over the course of a breeding program, highlights the importance of algorithm selection for the prediction of trait values.


Saturday, June 08, 2019
London: CogX, Founders Forum, Healthtech

I'm in London again to give the talk below and attend some meetings, including Founders Forum and their Healthtech event the day before.
CogX: The Festival of AI and Emerging Technology
King's Cross, London, N1C 4BH
When Machine Learning Met Genetic Engineering
3:30 pm Tuesday June 11 Cutting Edge stage
Speakers
Stephen Hsu
Senior Vice-President for Research and Innovation
Michigan State University
Helen O’Neill
Lecturer in Reproductive and Molecular Genetics
UCL
Martin Varsavsky
Executive Chairman
Prelude Fertility
Azeem Azhar (moderator)
Founder
Exponential View
Regent's Canal, Camden Town near King's Cross.




CogX speakers reception, Sunday evening:


HealthTech

Commanding heights of global capital:


Sunset, Camden locks:

Sunday, June 02, 2019
Genomic Prediction: Polygenic Risk Score for Type 1 Diabetes
In an earlier post I collected links related to recent progress in Polygenic Risk Scores (PRS) and health care applications. The paper below describes a new (published in 2019) predictor for Type 1 Diabetes (T1D) that achieves impressive accuracy (AUC > 0.9) using 67 SNPs. It incorporates model features such as nonlinear interactions between haplotypes.

T1D is highly heritable and tends to manifest at an early age. One application of this predictor is to differentiate between T1D and the more common (in later life) T2D. Another application is to embryo screening. Genomic Prediction has independently validated this predictor on sibling data and may implement it in their embryo biopsy pipeline, which includes tests for aneuploidy, single gene mutations, and polygenic risk.

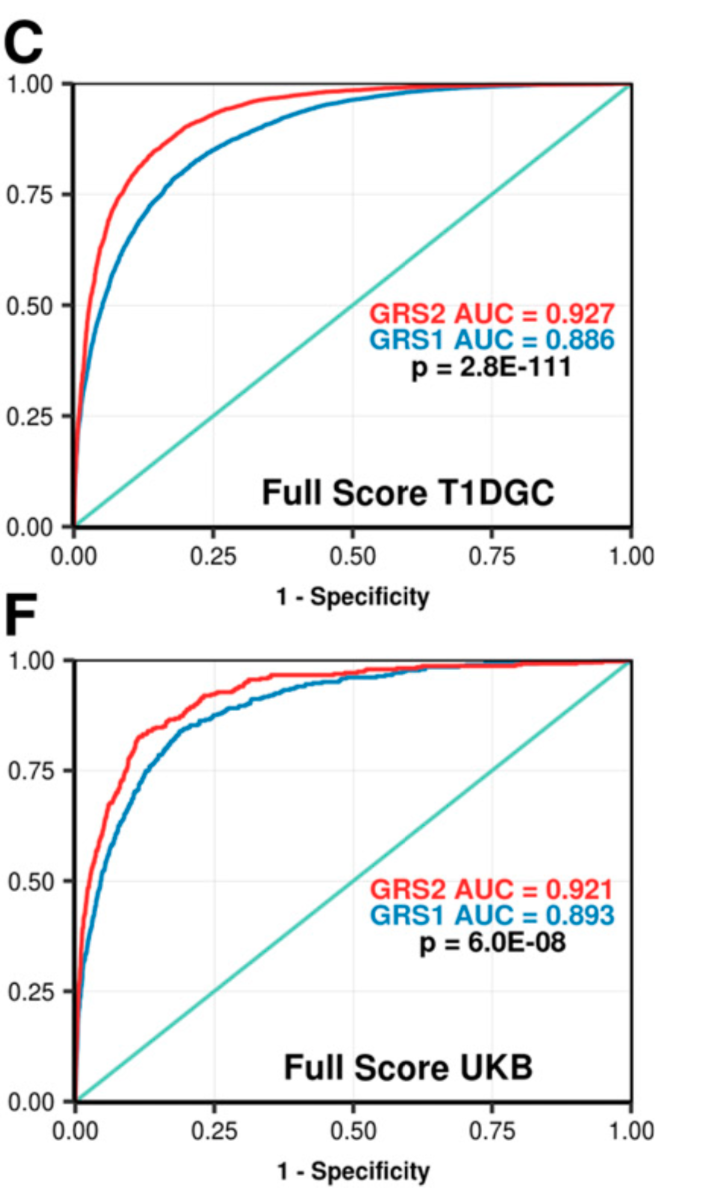
T1D is highly heritable and tends to manifest at an early age. One application of this predictor is to differentiate between T1D and the more common (in later life) T2D. Another application is to embryo screening. Genomic Prediction has independently validated this predictor on sibling data and may implement it in their embryo biopsy pipeline, which includes tests for aneuploidy, single gene mutations, and polygenic risk.
Development and Standardization of an Improved Type 1 Diabetes Genetic Risk Score for Use in Newborn Screening and Incident DiagnosisThe figure below gives some idea as to the ability of the new predictor GRS2 (panels B and D) to differentiate cases vs controls, and T1D vs T2D.
Sharp, et al.
Diabetes Care 2019;42:200–207 | https://doi.org/10.2337/dc18-1785
OBJECTIVE
Previously generated genetic risk scores (GRSs) for type 1 diabetes (T1D) have not captured all known information at non-HLA loci or, particularly, at HLA risk loci. We aimed to more completely incorporate HLA alleles, their interactions, and recently discovered non-HLA loci into an improved T1D GRS (termed the “T1D GRS2”) to better discriminate diabetes subtypes and to predict T1D in newborn screening studies.
RESEARCH DESIGN AND METHODS
In 6,481 case and 9,247 control subjects from the Type 1 Diabetes Genetics Consortium, we analyzed variants associated with T1D both in the HLA region and across the genome. We modeled interactions between variants marking strongly associated HLA haplotypes and generated odds ratios to create the improved GRS, the T1D GRS2. We validated our findings in UK Biobank. We assessed the impact of the T1D GRS2 in newborn screening and diabetes classification and sought to provide a framework for comparison with previous scores.
RESULTS
The T1D GRS2 used 67 single nucleotide polymorphisms (SNPs) and accounted for interactions between 18 HLA DR-DQ haplotype combinations. The T1D GRS2 was highly discriminative for all T1D (area under the curve [AUC] 0.92; P < 0.0001 vs. older scores) and even more discriminative for early-onset T1D (AUC 0.96). In simulated newborn screening, the T1D GRS2 was nearly twice as efficient as HLA genotyping alone and 50% better than current genetic scores in general population T1D prediction.
CONCLUSIONS
An improved T1D GRS, the T1D GRS2, is highly useful for classifying adult incident diabetes type and improving newborn screening. Given the cost-effectiveness of SNP genotyping, this approach has great clinical and research potential in T1D.

Subscribe to:
Posts (Atom)