
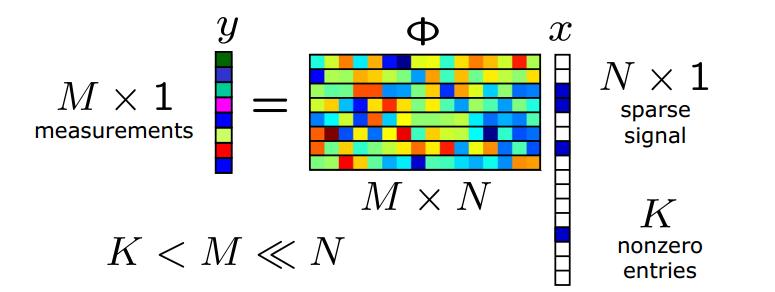
Compressed sensing (see also here) is a method for efficient solution of underdetermined linear systems: y = Ax + noise , using a form of penalized regression (L1 penalization, or LASSO). In the context of genomics, y is the phenotype, A is a matrix of genotypes, x a vector of effect sizes, and the noise is due to nonlinear gene-gene interactions and the effect of the environment. (Note the figure above, which I found on the web, uses different notation than the discussion here and the paper below.)
Let p be the number of variables (i.e., genetic loci = dimensionality of x), s the sparsity (number of variables or loci with nonzero effect on the phenotype = nonzero entries in x) and n the number of measurements of the phenotype (i.e., the number of individuals in the sample = dimensionality of y). Then A is an n x p dimensional matrix. Traditional statistical thinking suggests that n > p is required to fully reconstruct the solution x (i.e., reconstruct the effect sizes of each of the loci). But recent theorems in compressed sensing show that n > C s log p is sufficient if the matrix A has the right properties (is a good compressed sensor). These theorems guarantee that the performance of a compressed sensor is nearly optimal -- within an overall constant of what is possible if an oracle were to reveal in advance which s loci out of p have nonzero effect. In fact, one expects a phase transition in the behavior of the method as n crosses a critical threshold given by the inequality. In the good phase, full recovery of x is possible.
In the paper below, available on arxiv, we show that
1. Matrices of human SNP genotypes are good compressed sensors and are in the universality class of random matrices. The phase behavior is controlled by scaling variables such as rho = s/n and our simulation results predict the sample size threshold for future genomic analyses.
2. In applications with real data the phase transition can be detected from the behavior of the algorithm as the amount of data n is varied. A priori knowledge of s is not required; in fact one deduces the value of s this way.
3. For heritability h2 = 0.5 and p ~ 1E06 SNPs, the value of C log p is ~ 30. For example, a trait which is controlled by s = 10k loci would require a sample size of n ~ 300k individuals to determine the (linear) genetic architecture.
Application of compressed sensing to genome wide association studies and genomic selection
http://arxiv.org/abs/1310.2264
Authors: Shashaank Vattikuti, James J. Lee, Stephen D. H. Hsu, Carson C. Chow
Categories: q-bio.GN
Comments: 27 pages, 4 figures; Supplementary Information 5 figures
We show that the signal-processing paradigm known as compressed sensing (CS)
is applicable to genome-wide association studies (GWAS) and genomic selection
(GS). The aim of GWAS is to isolate trait-associated loci, whereas GS attempts
to predict the phenotypic values of new individuals on the basis of training
data. CS addresses a problem common to both endeavors, namely that the number
of genotyped markers often greatly exceeds the sample size. We show using CS
methods and theory that all loci of nonzero effect can be identified (selected)
using an efficient algorithm, provided that they are sufficiently few in number
(sparse) relative to sample size. For heritability h2 = 1, there is a sharp
phase transition to complete selection as the sample size is increased. For
heritability values less than one, complete selection can still occur although
the transition is smoothed. The transition boundary is only weakly dependent on
the total number of genotyped markers. The crossing of a transition boundary
provides an objective means to determine when true effects are being recovered.
For h2 = 0.5, we find that a sample size that is thirty times the number
of nonzero loci is sufficient for good recovery.
8 comments:
but what's n when the sample is drawn from extremes?
Is sparsity preferred because it is more biologically plausible than a non-sparse model?
Have you looked into using a Bayesian regression to achieve sparsity? Typically you would let each x_i follow a normal distribution x_i ~ Normal(x_i, mean, variance) but it is possible to instead have x_i ~ z * Normal(x_i, mean, variance) + (1 - z) * delta(x_i = 0). This is called a spike and slab model (with a slab defined by a normal distribution and a spike at zero) and it has a simple approximation based on expectation propagation.
Yes. It would be surprising if all loci affected a particular trait and also counter to initial indications from GWAS. Various priors like the one you suggested are similar to the prior of sparsity. But in the case of L1 penalization there are rigorous results like the ones we reference in our paper.
L1 penalization, a mixed finite continuous distribution.
this is typical.
the reality for phenomena outside the proper purview of physics will never correspond to any such absurdly simple models.
L1 penalization is just a method that allows one to efficiently find the sparsest solution. If you want to complain about idealization complain about the additive (linear) genetic model. But there are empirical justifications for this approximation.
Can you recommend a good reference discussing the empirical justifications for the additive linear approximation? I am especially interested in a discussion with some quantification of the relative effect sizes for specific examples (an added bonus would be something about genetic/environmental interactions).
I found this paper http://www.plosgenetics.org/article/info%3Adoi%2F10.1371%2Fjournal.pgen.1000008 to be useful in understanding why additive genetic variance makes sense in the real world.
Thanks, Aaron. Here are more relevant links.
http://infoproc.blogspot.com/2011/08/footnotes-and-citations.html
http://infoproc.blogspot.com/2013/02/eric-why-so-gloomy.html
http://infoproc.blogspot.com/2011/08/epistasis-vs-additivity.html
Post a Comment