
The Hsu BoundaryThere are many comments on Thompson's blog post, some of them confused. Comments from a user "Donoho-Student" are mostly correct -- he or she seems to understand the subject. (The phase transition discussed is related to the Donoho-Tanner phase transition. More from Igor Carron.)
... The “Hsu boundary” is Steve Hsu’s estimate that a sample size of roughly 1 million people may be required to reliably identify the genetic signals of intelligence.
... the behaviour of an optimization algorithm involving a million variables can change suddenly as the amount of data available increases. We see this behavior in the case of Compressed Sensing applied to genomes, and it allows us to predict that something interesting will happen with complex traits like cognitive ability at a sample size of the order of a million individuals.
Machine learning is now providing new methods of data analysis, and this may eventually simplify the search for the genes which underpin intelligence.
The chain of logic leading to this prediction has been discussed here before. The excerpt below is from a 2013 post The human genome as a compressed sensor:
For more posts on compressed sensing, L1-penalized optimization, etc. see here. Because s could be larger than 10k, the common SNP heritability of cognitive ability might be less than 0.5, and the phenotype measurements are noisy, and because a million is a nice round figure, I usually give that as my rough estimate of the critical sample size for good results. The estimate that s ~ 10k for cognitive ability and height originates here, but is now supported by other work: see, e.g., Estimation of genetic architecture for complex traits using GWAS data.
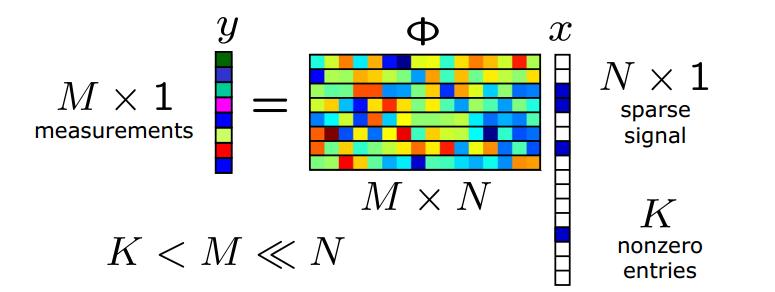
Compressed sensing (see also here) is a method for efficient solution of underdetermined linear systems: y = Ax + noise , using a form of penalized regression (L1 penalization, or LASSO). In the context of genomics, y is the phenotype, A is a matrix of genotypes, x a vector of effect sizes, and the noise is due to nonlinear gene-gene interactions and the effect of the environment. (Note the figure above, which I found on the web, uses different notation than the discussion here and the paper below.)
Let p be the number of variables (i.e., genetic loci = dimensionality of x), s the sparsity (number of variables or loci with nonzero effect on the phenotype = nonzero entries in x) and n the number of measurements of the phenotype (i.e., the number of individuals in the sample = dimensionality of y). Then A is an n x p dimensional matrix. Traditional statistical thinking suggests that n > p is required to fully reconstruct the solution x (i.e., reconstruct the effect sizes of each of the loci). But recent theorems in compressed sensing show that n > C s log p is sufficient if the matrix A has the right properties (is a good compressed sensor). These theorems guarantee that the performance of a compressed sensor is nearly optimal -- within an overall constant of what is possible if an oracle were to reveal in advance which s loci out of p have nonzero effect. In fact, one expects a phase transition in the behavior of the method as n crosses a critical threshold given by the inequality. In the good phase, full recovery of x is possible.
In the paper below, available on arxiv, we show that
1. Matrices of human SNP genotypes are good compressed sensors and are in the universality class of random matrices. The phase behavior is controlled by scaling variables such as rho = s/n and our simulation results predict the sample size threshold for future genomic analyses.
2. In applications with real data the phase transition can be detected from the behavior of the algorithm as the amount of data n is varied. A priori knowledge of s is not required; in fact one deduces the value of s this way.
3. For heritability h2 = 0.5 and p ~ 1E06 SNPs, the value of C log p is ~ 30. For example, a trait which is controlled by s = 10k loci would require a sample size of n ~ 300k individuals to determine the (linear) genetic architecture.
We have recently finished analyzing height using L1-penalization and the phase transition technique on a very large data set (many hundreds of thousands of individuals). The paper has been submitted for review, and the results support the claims made above with s ~ 10k, h2 ~ 0.5 for height.
Added: Here are comments from "Donoho-Student":
Donoho-Student says:
September 14, 2017 at 8:27 pm GMT • 100 Words
The Donoho-Tanner transition describes the noise-free (h2=1) case, which has a direct analog in the geometry of polytopes.
The n = 30s result from Hsu et al. (specifically the value of the coefficient, 30, when p is the appropriate number of SNPs on an array and h2 = 0.5) is obtained via simulation using actual genome matrices, and is original to them. (There is no simple formula that gives this number.) The D-T transition had only been established in the past for certain classes of matrices, like random matrices with specific distributions. Those results cannot be immediately applied to genomes.
The estimate that s is (order of magnitude) 10k is also a key input.
I think Hsu refers to n = 1 million instead of 30 * 10k = 300k because the effective SNP heritability of IQ might be less than h2 = 0.5 — there is noise in the phenotype measurement, etc.
Donoho-Student says:
September 15, 2017 at 11:27 am GMT • 200 Words
Lasso is a common statistical method but most people who use it are not familiar with the mathematical theorems from compressed sensing. These results give performance guarantees and describe phase transition behavior, but because they are rigorous theorems they only apply to specific classes of sensor matrices, such as simple random matrices. Genomes have correlation structure, so the theorems do not directly apply to the real world case of interest, as is often true.
What the Hsu paper shows is that the exact D-T phase transition appears in the noiseless (h2 = 1) problem using genome matrices, and a smoothed version appears in the problem with realistic h2. These are new results, as is the prediction for how much data is required to cross the boundary. I don’t think most gwas people are familiar with these results. If they did understand the results they would fund/design adequately powered studies capable of solving lots of complex phenotypes, medical conditions as well as IQ, that have significant h2.
Most ML people who use lasso, as opposed to people who prove theorems, are not aware of the D-T transition. Even most people who prove theorems have followed the Candes-Tao line of attack (restricted isometry property) and don’t think much about D-T. Although D eventually proved some things about the phase transition using high dimensional geometry, it was initially discovered via simulation using simple random matrices.













