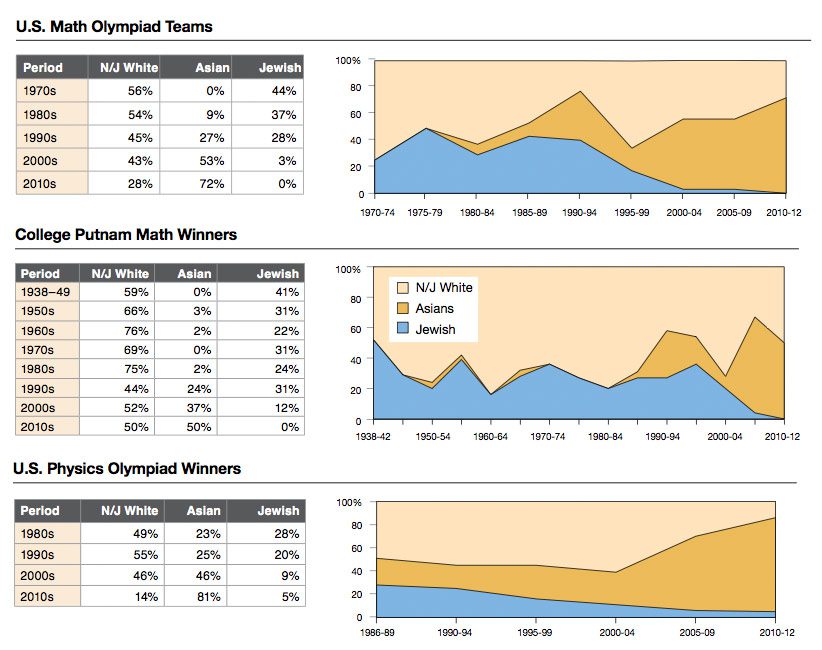
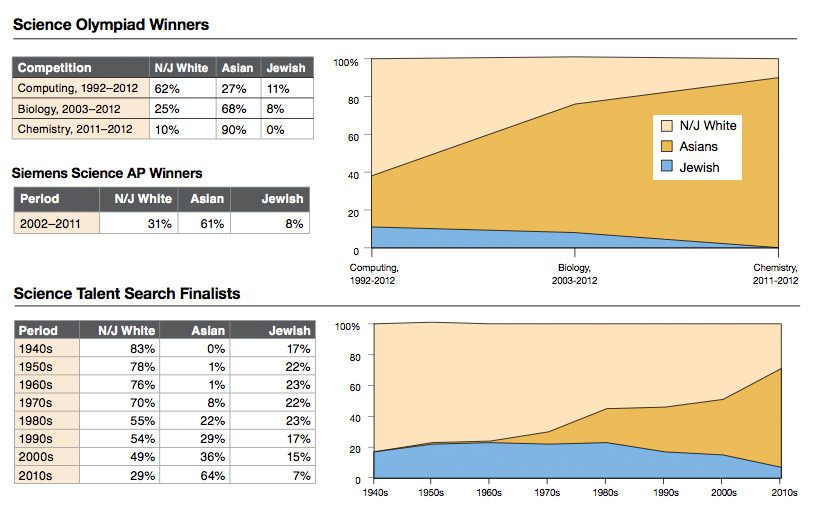
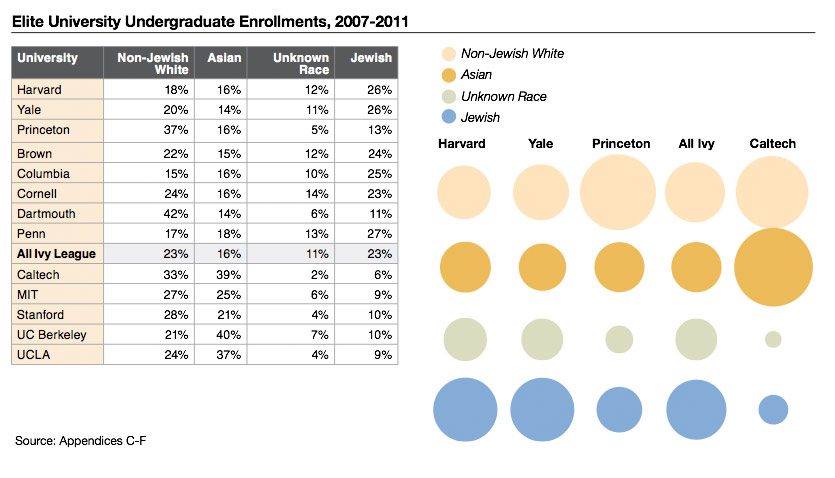
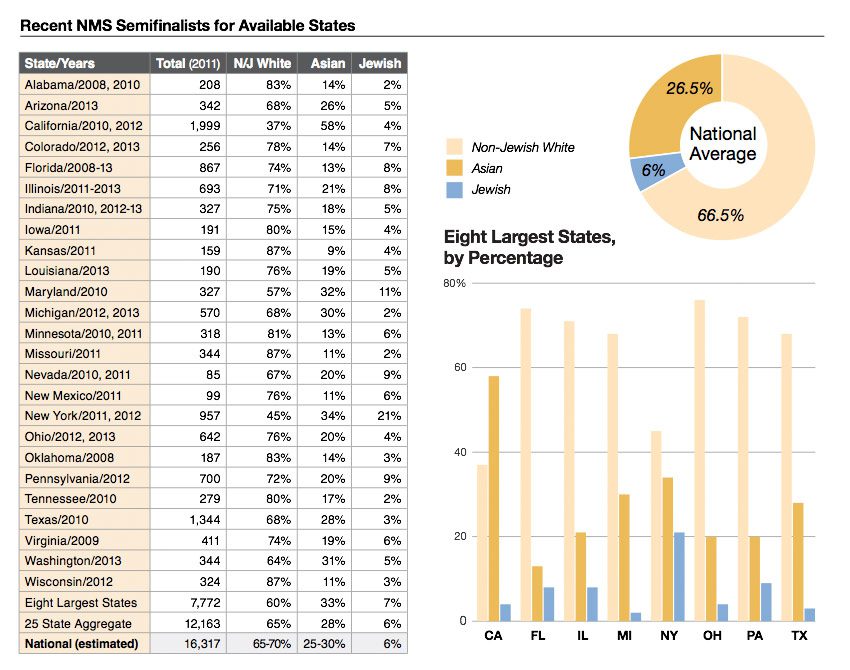
Eric Turkheimer wrote a
blog post reacting to my comments (
On the verge) about some recent intelligence GWAS results.
I'm an admirer of Eric's work in behavior genetics, as you can tell from this 2008 post
The joy of Turkheimer. Since then we've gotten to know each other via the internet and have even met at a
conference.
Eric is famous for (among other things) his Gloomy Prospect:
The question is not whether there are correlations to be found between individual genes and complex behavior— of course there are — but instead whether there are domains of genetic causation in which the gloomy prospect does not prevail, allowing the little bits of correlational evidence to cohere into replicable and cumulative genetic models of development. My own prediction is that such domains will prove rare indeed, and that the likelihood of discovering them will be inversely related to the complexity of the behavior under study.
He is right to be cautious about whether discovery of individual gene-trait associations will cohere into a satisfactory explanatory or predictive framework. It is plausible to me that the workings of the DNA program that
creates a human brain are incredibly complex and beyond our detailed understanding for some time to come.
However, I am optimistic about the prediction problem. There are good reasons to think that the linear term in the model described below gives the dominant contribution to variation in cognitive ability:
The evidence comes from estimates of
additive (linear) variance in twin and adoption studies, as well as from evolutionary theory itself. Fisher's Fundamental Theorem of Natural Selection identifies
additive variance as the main driver of evolutionary change in the limit where selection timescales are much longer than recombination (e.g., due to sexual reproduction) timescales. Thus it is reasonable to expect that most of the change in genus Homo intelligence over the last millions of years is encoded in a linear genetic architecture.
GWAS, which identify causal loci and their effect sizes, are in fact fitting the parameters of the linear model that appears in the slide above. (Most effect sizes x_i will be zero, with perhaps 10k non-zero entries distributed according to some kind of power law.) Once we have characterized loci accounting for most of the variance, we will be able to predict phenotypes based only on genotype information (i.e., without further information about the individual). This is the genomic prediction problem which has already been
partially solved for inbred lines of domesticated plants and animals. My guess is that it will be solved for humans once of order millions of genotype-phenotype pairs are available for analysis. Understanding the nonlinear parts will probably take much more data, but these are likely to be subleading effects.